Wir haben diesen Beitrag im laufenden Monat überprüft und die Beschreibungen teilweise aktualisiert.
Unsere Empfehlungen sind nach wie vor auf dem neuesten Stand. Letztes Updated am 29. September 2020
Was sind Hämolytische Anämien?
Neben angeborenen Formen finden sich erworbene hämolytische Anämien. Haptoglobin ist bei intravasalen Hämolysen schneller erniedrigt als bei extravasalen Hämolysen. Die Retikulozytenzahl gibt einen Hinweis auf die vorliegende Anämie (erniedrigt bei Störungen der Knochenmarkregenerationsfähigkeit, erhöht bei peripherem Abbau der Erythrozyten).
Auf einen Blick: Das Wichtigste in Kürze
[su_icon_text color=“#333333″ icon=“icon: check-square-o“ icon_color=“#afafaf“ icon_size=“40″ url=““ target=“blank“ class=““]Die meisten pathologischen Hämolysen sind extravasal und treten auf, wenn geschädigte oder abnorme Erythrozyten durch die Milz und die Leber aus dem Blutkreislauf entfernt werden.[/su_icon_text] [su_icon_text color=“#333333″ icon=“icon: check-square-o“ icon_color=“#afafaf“ icon_size=“40″ url=““ target=“blank“ class=““]Der normale Abbau überalterter roter Blutkörperchen (Erythrozyten) erfolgt hauptsächlich durch die Makrophagen.[/su_icon_text] [su_icon_text color=“#333333″ icon=“icon: check-square-o“ icon_color=“#afafaf“ icon_size=“40″ url=““ target=“blank“ class=““]Hämolytische Anämien sind durch die intravasale oder extravasale Zerstörung von Erythrozyten gekennzeichnet. Sie werden manifest, wenn die Produktion der Erythrozyten im Knochenmark langsamer verläuft als deren Abbau[/su_icon_text]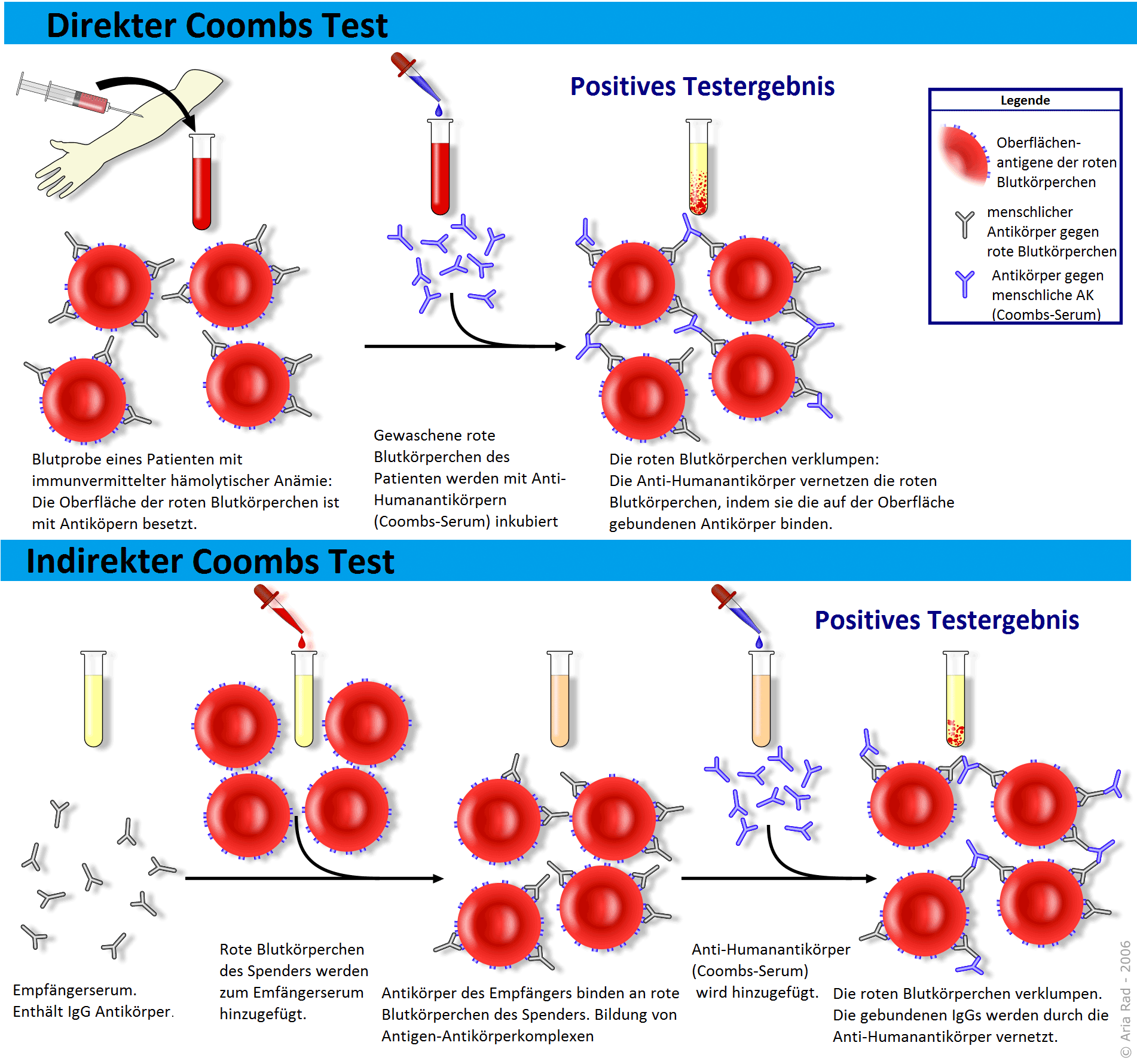
Angeborene hämolytische Anämien
Unter den angeborenen hämolytischen Anämien unterscheidet man korpuskuläre Formen von nichtkorpuskulären Formen.
Kugelzellenanämie: Eine häufige hämolytische Anämie ist die Kugelzellenanämie (Sphärozytose). Der Name beruht auf der Eigenschaft der Erythrozyten, eine kugelige Form einzunehmen. Das MCV ist normal. Hingegen ist das MCH erhöht (> 36 g/dl).
Ursächlich liegt ein Defekt der Erythrozytenmembran vor (entweder ein isolierter Spektrinmangel, häufiger ein kombinierter Spektrin-/Ankyrinmangel). Dies führt zu einer Störung der Ionenpermeabilität mit Natrium- und Wassereinstrom in die Erythrozyten und Kugelformbildung. Im peripheren Ausstrich sieht man über 50 % Mikrosphärozyten. Die Diagnose wird über die Bestimmung der osmotischen Resistenz der Erythrozyten gesichert. Diese ist bei der Erkrankung immer vermindert. Die Hämolyse tritt bereits bei NaCl-Konzentrationen über 0,5% ein. Da auch bei einer AIHA die osmotische Resistenz vermindert sein kann, wird zur Differenzierung der Coombs-Test bestimmt, der bei der Sphärozytose negativ ist. Patienten mit Kugelzellenanämie weisen i. d. R. einen Subikterus ohne klinische Symptome auf. Probleme bereiten im Verlauf der Erkrankung die Gallensteine, die aufgrund der chronischen Hämolyse entstehen. Im Rahmen von Virusinfektionen kann es zu hämolytischen Krisen kommen.
Thalassämie und Sichelzellenanämie
Thalassämie: Die Thalassämie wurde bereits unter den hypochromen Anämien besprochen.
Sichelzellenanämie: Bei der autosomal-rezessiven Sichelzellenanämie liegt ein strukturell verändertes Hb vor, in dessen ß-Ketten an Position 6 Glutaminsäure gegen Valin ausgetauscht ist. Dieses abnorme Globin kann in der Hb-Elektro- phorese als HbS nachgewiesen werden. Die Sichelbildung der Erythrozyten entsteht bei niedrigem Sauerstoffpartialdruck im Blut und führt zur Polymerisation des abnormen Hb. Dies wiederum führt zur Rigidität und Unverformbarkeit der Erythrozyten mit der Folge einer Okklusion der Kapillaren, die zu Schmerzkrisen führt. Eine Splenomegalie ist nicht die Norm, da durch Autosequestration häufig eine Milzatrophie resultiert, worauf Jolly-Körperchen im Ausstrich hinweisen. Die Patienten weisen eine Retikulozytose auf. Nur homozygote Träger erkranken. Bei heterozygoten Trägern lassen sich Sichelzellen nur im sauerstoffarmen Milieu nachweisen (Sichelzellen-Test). Dies hat klinisch keine Relevanz, die Träger sind gesund.

Merke: Bel Verdacht auf Vorliegen angeborener, mit Hämolyse einhergehender Erkrankungen sollte eine Vorstellung in einem speziellen Zentrum veranlasst werden. Die Sichelzellenanämie sollte aber durch jeden Arzt im Differentialblutbild erkannt werden.
Glukose-6-Phosphat-Dehydrogenasemangel (G-6-PD-Mangel, Favismus):
Der G-6-PD-Mangel ist eine häufige, X-chromosomal-rezessiv vererbte Erkrankung. Bei den Betroffenen (Afrikaner, Asiaten, Personen aus dem Mittelmeerraum) sind über 300 Mutationen beschrieben. Männer erkranken immer, homozygote Frauen können gesund oder krank sein. Es besteht eine erhöhte Resistenz gegen Infektionen mit Plasmodien. Der G-6-PD-Mangel führt zur verminderten Bildung von reduziertem Glutathion. Durch oxidativen Stress (Infektionen, Arzneimittel) entstehen Peroxide, die nicht entgiftet werden können und zur Erythrozytenschädigung führen. Heinz-Innenkörper können im Schub nachgewiesen werden (Denaturierungsprodukte des Hb). Beweisend ist der Nachweis der verminderten G-6-PD-Aktivität in den Erythrozyten.
Pyruvatkinasemangel (PK-Mangel): Bei dem autosomal-rezessiv vererbten PK- Mangel ist die Glykolyse der Erythrozyten gestört. Nur homozygote Träger erkranken mit einer hämolytischer Anämie und häufig auch einer Splenomegalie. Da den Erythrozyten zur Energiegewinnung nur die Glykolyse zur Verfügung steht, führt ein PK-Mangel zu einem ATP-Mangel, Störungen der Na*/K*-ATPase der Erythrozytenmembran und damit zu einer Membraninstabilität mit der Folge einer Hämolyse. Meist sind die Patienten asymptomatisch. Bei homozyoten Trägern können hämolytische Krisen auftreten. Im Blutausstrich lassen sich Akanthozyten nachweisen. Beweisend ist die Bestimmung einer verminderten PK-Aktivität in den Erythrozyten.
Quellen:
- https://www.msdmanuals.com/de-de/profi/h%C3%A4matologie-und-onkologie/h%C3%A4molytische-an%C3%A4mien/h%C3%A4molytische-an%C3%A4mien-im-%C3%BCberblick
- https://www.netdoktor.at/krankheit/haemolytische-anaemie-7344
- https://www.lecturio.de/magazin/haemolytische-anaemien/
Ständig neue Beiträge
Wir bieten hier fachlich geprüfte Gesundheitsinformationen, die in allgemein verständlicher Sprache verfasst sind. Wir zeigen ausführliche Informationen zu Blutwerten, Laborwerten und beschreiben Krankheiten und deren Symptome, teils gibt es auch Therapiemöglichkeiten. Die textlichen und grafischen Inhalte dieses Portals werden ständig erweitert, sodass Sie hier und auf unserer Facebook Seite viele Gesundheitsinformationen finden.
• Unsere redaktionelle Qualitätssicherung • Beratung und Hilfe

